
| Research | |
|
Squeezing and shearing behaviors of nonpolar liquid films in nanoscale confined geometry
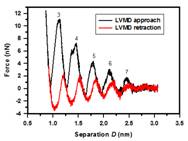
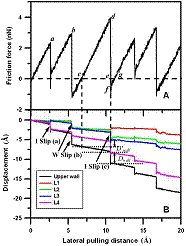
The physical property of liquid film confined between two surfaces to nanometers thickness is of fundamental interest in surface and interfacial science. How the film reacts to external loading in a confined geometry is relevant to friction and lubrication. Many surface force apparatus (SFA) or surface force balance (SFB) experiments found that the solvation force versus distance always exhibits force oscillations within a distance of 7-8 molecular diameters of the liquid molecules. However, the nature of this oscillatory force and layering transition is still not quite understood. We have recently developed a liquid-vapor molecular dynamics (LVMD) simulation method to study this interesting phenomenon. Our findings through normal approach and retraction simulations for a simple liquid argon show that the layering transition is an abrupt, liquid-to-solid (solidification) and then solid-to-liquid (melting) phase transition process. During the solidification, the nucleation of solid phase initiates in the central region and spread outward that can support a finite shear stress, while during the melting, there is an inward shrinking of the solid phase. The speed of the squeeze out front (the solid-liquid phase boundary) is observed in the order of a few m/s. But what about the friction behavior of solidified film? Usually stick-slip motion of solids over each other will occur. This is often observed in our daily lives. A common knowledge to explain this phenomenon is associated with the crystallization and shear melting of the confined film. During the stick, a finite shear stress or static friction force is built up within the solidified film. When the maximum shear stress within the film is exceeded, shear melting of the film occurs, resulting in the slip motion of solids. This slip proceeds until the recrystallization of the film begins. However, our studies show that, instead of shear melting and recrystallization of the film, interlayer slips within the solidified film and wall slips at the wall-film interface are often observed. The ordered solidified film is well maintained during the slip. Through the time variations of the frictional force and potential energy change within the film, we find that both the friction dissipation during the slip and the potential energy decay after the slip in the solidified film take a fairly large portion of the total energy dissipation. Publications: 1. Xu, R. G. and Leng, Y. S., "Solvation force simulations in atomic force microscopy," J. Chem. Phys., 140, 214702 (2014) |

|
Hydration force and hydrophobic interactions in aqueous system
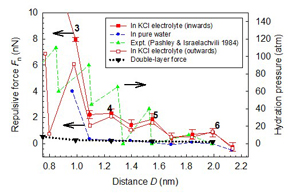
The mechanical properties of aqueous films under nanometers confinement have many implications in materials science and engineering. Adhesion and wetting, friction and lubrication in micro/nano electro-mechanical systems (M/NEMS) are among typical examples. Many biological processes, such as interactions between biomembranes, water transport through ion channels and through crowded intracellular environment, protein folding and biolubrication, all involve water-hydrophobic or water-hydrophilic group interactions, and in many cases hydrated metal ions are present that play a significant role. Our recent studies using LVMD simulation reveal the repulsive hydration force mechanism between two mica surfaces in KCl electrolyte. Strong repulsive hydration force was observed within a distance of 2 nm between two charged mica surfaces, which cannot be explained by the continuum theory of double-layer repulsion. We attribute the load bearing capacity of the dense electrolyte to the very hard hydration shells of K+ metal ions under confinement. We find that the hydrated K+ ions and Cl− co-ions remain very diffusive in the aqueous film. Water molecules in the hydration layer are also very fluidic, in the sense that the diffusion constant of water molecules is less than its bulk value by at most three orders of magnitude even under subnanometer extreme confinement. Hydrophobic drying and hysteresis is an interesting topic related to protein folding and many hydrophobic interactions. For two parallel hydrophobic plates immersed in water, we find that there is a length scale effect on the hydrophobic drying during normal approach and the hydrophobic hysteresis during retraction. The critical distance at which a spontaneous drying (hydrophobic collapse) occurs is determined by the shorter characteristic dimension of the plate, while the hydrophobic hysteresis is determined by the longer characteristic dimension of the plate. The variations of the potential of mean force versus separation during approach and retraction show that water confined between two parallel hydrophobic plates is in a thermodynamic metastable state. This comparably high energy state leads to the spontaneous drying at the critical distances. Publications: 1. Leng, Y. S., “Hydration force between mica surfaces in aqueous KCl electrolyte solution,” Langmuir 28, 5339 (2012)2. Lei, Y. J. and Leng, Y. S., “Hydrophobic drying and hysteresis at different length scales by molecular dynamics simulations,” Langmuir 28, 3152 (2012) 3. Leng, Y. S., "Hydration force and dynamic squeeze out of hydration water under subnanometer confinement," Journal of Physics: Condensed Matter 20, 354017(2008) |

|
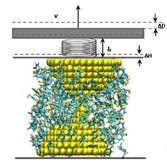
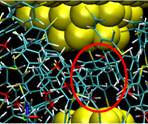
Driven dynamics of single molecular junctions
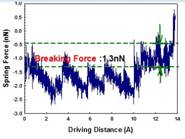
The formation and mechanical properties of single molecular junctions are fundamental problems in molecular electronics devices. The conformation of metal-molecule-metal junctions and their dynamics evolution during pulling are still not quite understood. Using a driven dynamics model, we studied the stable structures of single atomic contacts in gold nanojunctions and the formation of gold-molecule-gold junctions in dithiol solvent. Through repeated jump-to-contact and jump-out-of-contact trainings we identified three stable elastic contacts in gold nanojunctions. The breaking force calculated is consistent with the mechanically controllable break junction (MCBJ) experimental result. Using a combination of grand canonical Monte Carlo and molecular dynamics simulations, we simulate the pulling of gold nanojunction in benzenedithiol-toluene solvent. We find that the gold-thiolate binding is much stronger than the gold-gold bond. Consequently, the breakdown happens at the gold-gold bond, rather than at the gold-sulfur bond. Publications: 1. Wang, H. C. and Leng, Y. S., “Molecular dynamics simulations of the stable structures of single atomic contacts in gold nanojunctions,” Phys. Rev. B 84, 245422 (2011) |
|
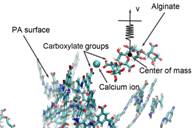
Membrane fouling in water purification
Membrane fouling is a serious problem to negatively influence drinking water purification. To understand membrane-foulant interactions, we have performed experimental measurements (in collaboration with Baoxia Mi’s group at University of Maryland) and molecular simulations to calibrate binding characteristics between polyamide membrane and alginate molecules (one of the typical foulants in natural environment). Our studies show that hydrated metal ions play a central role in building ionic bridges to promote membrane fouling. In particular, Ca2+ ion has a stronger binding than Na+ ion, but with a shorter period of time for breakdown. Publications: 1. Xiang, Y., Liu, Y. L., Mi, B. X., and Leng, Y. S., "Molecular dynamics simulations of polyamide membrane, calcium alginate gel, and their interactions in aqueous solution," Langmuir, 30, 9098 (2014)2. Xiang, Y., Liu, Y. L., Mi, B. X., and Leng, Y. S., "Hydrated polyamide membrane and its interaction with alginate: A molecular dynamics study," Langmuir, 29, 11600 (2013) |

|
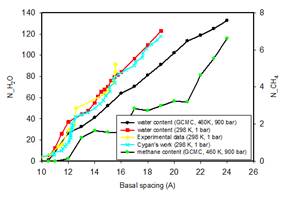
Clay swelling in aqueous-hydrocarbon system
Methane hydrate is abundant under sediments on the ocean floors and in the underground sedimentary basin. However clay swelling frequently results in formation damage of drilling well bore. While it is well known that water molecules can easily penetrate into interlayer of Na-montmorillonite to induce clay swelling, it is not clear how the methane hydrate plays a role in the swelling process. We have carefully calculated the chemical potentials for water and methane at different temperatures and pressures, corresponding to different burial depths underground, and then perform grand canonical Monte Carlo simulation to see how likely methane can form hydrate in clay interlayer. We found that at burial depth of 6 km, corresponding to 460 K and 900 bar pressure, methane can penetrate clay interlayer even at one hydration layer. As clay continues to swell, significant amount of methane hydrates will form in Na-montmorillonite clay. Methane hydrate can form inner-sphere structures with clay surface, as well as outer-sphere structures in thicker hydration layers. Publications: 1. Rao, Q. and Leng, Y. S., "Methane aqueous fluids in montmorillonite clay interlayer under near-surface geological conditions: A grand canonical Monte Carlo and molecular dynamics simulation study," J. Phys. Chem. B, 118, 10956 (2014)2. Rao, Q., Xiang, Y., and Leng, Y. S., "Molecular simulations on the structure and dynamics of water-methane fluids between Na-Montmorillonite clay surfaces at elevated temperature and pressure," J. Phys. Chem. C, 117, 14061 (2013) |
 |
Metal-semiconductor contact in sliding nanogenerator
There have been significant progresses in the development of high-power, ZnO-based piezoelectric nanogenerators. However, there is also a compelling need to understand, at atomistic level, the structural and dynamic properties of the noble metal-ZnO interface. These properties are critical to the performance of the devices, in the sense that they are closely related to the electron transport and the mechanical stability of the metal–semiconductor contacts. We study Pt–ZnO sliding contact and piezoelectric potential distribution for a model system, and find that when a Pt (111) tip sliding over a (0001) end surface of the ZnO nanowire, from the tensile side to the compressive side during its lateral bending, shearing off one monolayer of Pt atoms can easily proceed, resulting in subsequent relaxations of Pt metal tip. However, no atomic interpenetration or material transfer between the metal tip and the semiconductor material occurs. The calculated piezoelectric potential distribution due to the bending of ZnO nanowire is similar to the result from the continuum theory calculations, but shows more detailed features at the interface. Publications: |
Copyright© 2014 Computational Materials Science Laboratory. All Rights Reserved